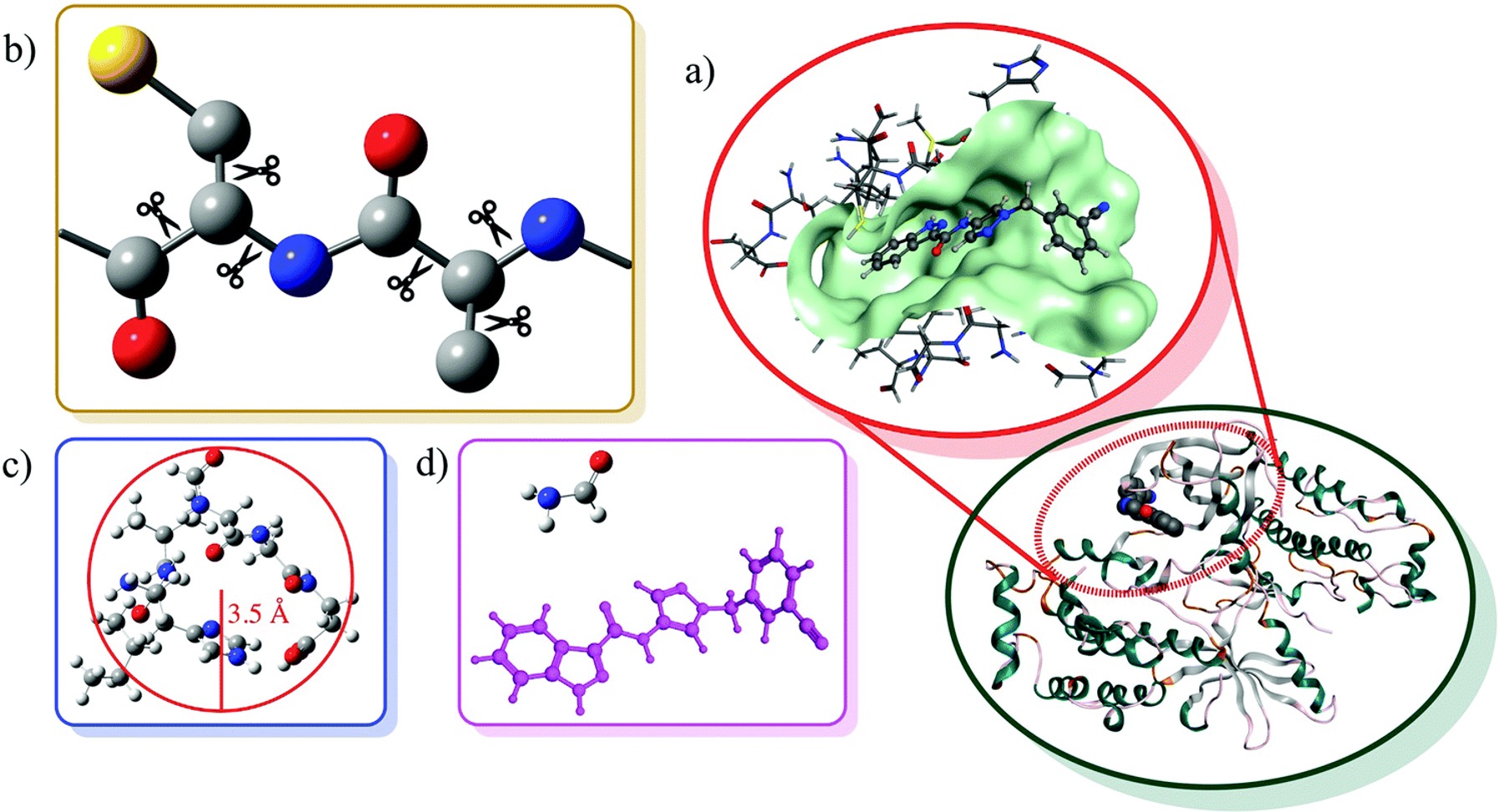
To address the steep computational scaling of our most accurate theoretical methods, we have recently developed a new extrapolated fragment-based approach, termed molecules-in-molecules (MIM), for accurate energy calculations on large molecules (ex: proteins). In this method, we use a multilevel partitioning approach coupled with electronic structure studies at multiple levels of theory to provide a hierarchical strategy for systematically improving the computed results.
In particular, we use a generalized hybrid energy expression, similar in spirit to that in the popular ONIOM methodology, that can be combined easily with any fragmentation procedure.
In the current work, we explore a MIM scheme which first partitions a molecule into non-overlapping fragments and then recombines the interacting fragments to form overlapping subsystems. By including all interactions with a cheaper level of theory, the MIM approach is shown to significantly reduce the errors arising from a single level fragmentation procedure. This is a very new and active field of research, with many exciting avenues to explore, both on the application and development fronts.
Recent Publications:
“Towards Post-Hartree-Fock Accuracy for Protein-Ligand Affinities Using the Molecules-in-Molecules Fragmentation-Based Method”, A. Gupta, S. Maier, B. Thapa, and K Raghavachari.(2024).https://doi/10.1021/acs.jctc.3c01293
“Comparative Assessment of QM-based and MM-based Models for Prediction of Protein-Ligand Binding Affinity Trends”, S. Maier, B. Thapa, J. Erickson and K. Raghavachari, Phys. Chem. Chem. Phys. 24, 14525–14537 (2022). https://doi.org/10.1039/d2cp00464j
“Exploring Protein-Ligand Interactions using the Multilayer Molecules-in-Molecules (MIM) Fragmentation-Based Approach”, B. Thapa, D. Beckett, J. Erickson, and K. Raghavachari, J. Chem. Theory Comput. 14, 5143-5155 (2018). http://dx.doi.org/10.1021/acs.jctc.8b00531